Vignette#
Example workflow to convert CITESeq data to FCS file format.
This example used to 5k PBMC NextGEM dataset from 10X.
Download data from: http://cf.10xgenomics.com/samples/cell-exp/3.1.0/5k_pbmc_protein_v3_nextgem/5k_pbmc_protein_v3_nextgem_filtered_feature_bc_matrix.h5
[1]:
import os
import scanpy as sc
from anndata_fcs import anndata_to_fcs, fcs_to_anndata, fcs_to_dataframe, gate_polygon, scatter, gate_polygon_subset
Load anndata object from rawdata#
[2]:
adata = sc.read_10x_h5(
filename=os.path.join(os.getcwd(), "..", "..", "data", "5k_pbmc_protein_v3_nextgem_filtered_feature_bc_matrix.h5"),
gex_only=False,
)
adata.var_names_make_unique()
/home/malte/Dokumente/Github/citeseq_to_fcs/venv/lib/python3.10/site-packages/anndata/_core/anndata.py:1758: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
Filter CITESeq data#
[3]:
adata_citeseq = adata[:, adata.var["feature_types"] == "Antibody Capture"]
adata_citeseq
[3]:
View of AnnData object with n_obs × n_vars = 5527 × 32
var: 'gene_ids', 'feature_types', 'genome', 'pattern', 'read', 'sequence'
Generate FCS file#
[4]:
# Convert data
fcs_data = anndata_to_fcs(adata_citeseq)
[5]:
adata_citeseq.obs.head()
[5]:
| barcode_rank | |
|---|---|
| AAACCCACAGGCTTGC-1 | 0 |
| AAACCCAGTAGTTAGA-1 | 1 |
| AAACGAAGTAACGATA-1 | 2 |
| AAACGAAGTGGATCAG-1 | 3 |
| AAACGAATCATGAGAA-1 | 4 |
[6]:
# Save fcs file
fcs_data.write_fcs(os.path.join(os.getcwd(), "..", "..", "data", "citeseq.fcs"))
[7]:
fcs_df = fcs_to_dataframe(fcs_data)
fcs_df.head()
[7]:
| CD3_TotalSeqB | CD4_TotalSeqB | CD8a_TotalSeqB | CD11b_TotalSeqB | CD14_TotalSeqB | CD15_TotalSeqB | CD16_TotalSeqB | CD19_TotalSeqB | CD20_TotalSeqB | CD25_TotalSeqB | ... | CD274_TotalSeqB | CD278_TotalSeqB | CD335_TotalSeqB | PD-1_TotalSeqB | HLA-DR_TotalSeqB | TIGIT_TotalSeqB | IgG1_control_TotalSeqB | IgG2a_control_TotalSeqB | IgG2b_control_TotalSeqB | barcode_rank | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 12.0 | 280.0 | 9.0 | 3122.0 | 746.0 | 9.0 | 1.0 | 5.0 | 2.0 | 4.0 | ... | 2.0 | 6.0 | 8.0 | 4.0 | 101.0 | 4.0 | 5.0 | 2.0 | 4.0 | 0.0 |
| 1 | 24.0 | 231.0 | 11.0 | 1241.0 | 355.0 | 10.0 | 2.0 | 4.0 | 11.0 | 5.0 | ... | 1.0 | 4.0 | 8.0 | 8.0 | 1450.0 | 5.0 | 0.0 | 1.0 | 1.0 | 1.0 |
| 2 | 23.0 | 117.0 | 3.0 | 582.0 | 133.0 | 7.0 | 4.0 | 1.0 | 7.0 | 3.0 | ... | 4.0 | 5.0 | 2.0 | 2.0 | 524.0 | 2.0 | 1.0 | 4.0 | 2.0 | 2.0 |
| 3 | 8.0 | 83.0 | 1.0 | 1966.0 | 675.0 | 7.0 | 2.0 | 3.0 | 8.0 | 3.0 | ... | 3.0 | 2.0 | 2.0 | 5.0 | 216.0 | 0.0 | 5.0 | 1.0 | 3.0 | 3.0 |
| 4 | 726.0 | 1100.0 | 5.0 | 14.0 | 13.0 | 6.0 | 2.0 | 7.0 | 6.0 | 11.0 | ... | 2.0 | 129.0 | 4.0 | 5.0 | 9.0 | 5.0 | 1.0 | 3.0 | 4.0 | 4.0 |
5 rows × 33 columns
Gating on FCS file#
[8]:
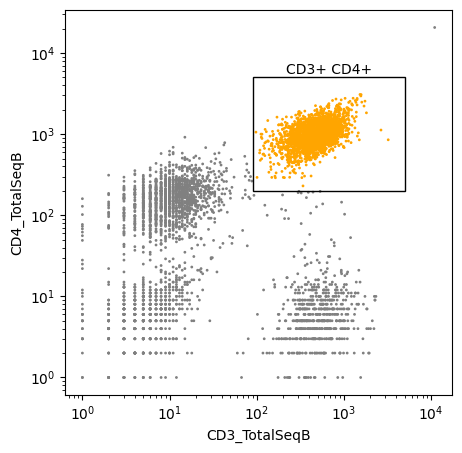
in_polygon = gate_polygon(
fcs_data,
x="CD3_TotalSeqB",
y="CD4_TotalSeqB",
polygon=[[90, 200], [5000, 200], [5000, 5000], [90, 5000]],
)
[9]:
ax = scatter(
data=fcs_df,
x="CD3_TotalSeqB",
y="CD4_TotalSeqB",
gates={"CD3+ CD4+": [[90, 200], [5000, 200], [5000, 5000], [90, 5000]]},
highlight=in_polygon,
highlight_color="orange",
color="gray",
)
[10]:
fcs_to_dataframe(
gate_polygon_subset(
fcs_data,
x="CD3_TotalSeqB",
y="CD4_TotalSeqB",
polygon=[[90, 200], [5000, 200], [5000, 5000], [90, 5000]],
)
).head()
[10]:
| CD3_TotalSeqB | CD4_TotalSeqB | CD8a_TotalSeqB | CD11b_TotalSeqB | CD14_TotalSeqB | CD15_TotalSeqB | CD16_TotalSeqB | CD19_TotalSeqB | CD20_TotalSeqB | CD25_TotalSeqB | ... | CD274_TotalSeqB | CD278_TotalSeqB | CD335_TotalSeqB | PD-1_TotalSeqB | HLA-DR_TotalSeqB | TIGIT_TotalSeqB | IgG1_control_TotalSeqB | IgG2a_control_TotalSeqB | IgG2b_control_TotalSeqB | barcode_rank | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 726.0 | 1100.0 | 5.0 | 14.0 | 13.0 | 6.0 | 2.0 | 7.0 | 6.0 | 11.0 | ... | 2.0 | 129.0 | 4.0 | 5.0 | 9.0 | 5.0 | 1.0 | 3.0 | 4.0 | 4.0 |
| 1 | 342.0 | 1086.0 | 2.0 | 9.0 | 10.0 | 4.0 | 1.0 | 3.0 | 7.0 | 11.0 | ... | 5.0 | 44.0 | 3.0 | 5.0 | 14.0 | 12.0 | 0.0 | 2.0 | 4.0 | 5.0 |
| 2 | 125.0 | 1144.0 | 1.0 | 26.0 | 8.0 | 8.0 | 2.0 | 2.0 | 5.0 | 17.0 | ... | 4.0 | 36.0 | 0.0 | 4.0 | 11.0 | 2.0 | 3.0 | 2.0 | 1.0 | 7.0 |
| 3 | 538.0 | 1065.0 | 3.0 | 11.0 | 15.0 | 5.0 | 3.0 | 2.0 | 6.0 | 2.0 | ... | 3.0 | 44.0 | 2.0 | 12.0 | 9.0 | 0.0 | 3.0 | 0.0 | 1.0 | 9.0 |
| 4 | 224.0 | 531.0 | 1.0 | 11.0 | 4.0 | 2.0 | 2.0 | 2.0 | 1.0 | 12.0 | ... | 0.0 | 14.0 | 1.0 | 2.0 | 9.0 | 2.0 | 1.0 | 1.0 | 4.0 | 10.0 |
5 rows × 33 columns
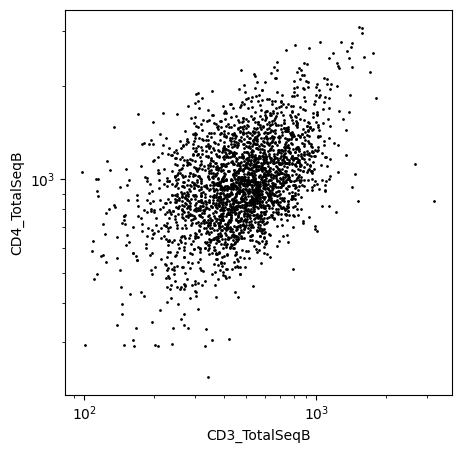
[11]:
ax = scatter(
data=fcs_to_dataframe(
gate_polygon_subset(
fcs_data,
x="CD3_TotalSeqB",
y="CD4_TotalSeqB",
polygon=[[90, 200], [5000, 200], [5000, 5000], [90, 5000]],
)
),
x="CD3_TotalSeqB",
y="CD4_TotalSeqB",
)
Recover anndata from gated FCS file#
[12]:
adata_output = fcs_to_anndata(
gate_polygon_subset(
fcs_data,
x="CD3_TotalSeqB",
y="CD4_TotalSeqB",
polygon=[[90, 200], [5000, 200], [5000, 5000], [90, 5000]],
),
include_metadata=False,
)
adata_output
[12]:
AnnData object with n_obs × n_vars = 2581 × 33
[13]:
adata_citeseq[adata_citeseq.obs["barcode_rank"].isin(adata_output[:, ["barcode_rank"]].X.flatten()), :]
[13]:
View of AnnData object with n_obs × n_vars = 2581 × 32
obs: 'barcode_rank'
var: 'gene_ids', 'feature_types', 'genome', 'pattern', 'read', 'sequence'